
Диагностика заболевания
Дисплазия слизистой не болит, не мешает жить – у неё нет симптомов.
Самый простой способ выявления патологии слизистой шейки матки был придуман Папаниколау в 1940-х годах и состоял он из взятия соскоба поверхностных клеток. Сегодня применяется модифицированный инструментарий, позволяющий собрать больше материала. Исследование клеток под микроскопом – цитология позволяет определиться во следующим диагностическим этапом – кольпоскопией.
Расширенная кольпоскопия – осмотр тканей под большим увеличением от пятикратного до 30-кратного, с дополнительным усилением «картинки» специальными обработками растворами, что помогает выбору оптимального места для взятия кусочка ткани – биопсии участка дисплазии. Кусочки слизистой размером не менее 3 миллиметров отправляются на микроскопию – гистологию. Биопсия исключается при воспалении и инфекциях, но только на время.

Дальше при морфологическом подтверждении дисплазии проводится выскабливание слизистой оболочки цервикального канала для выявления его изменений, у женщины дисплазия может локализоваться в железистых криптах – ямках слизистой и зона эпителиального перехода способна смещаться выше. Выскабливание визуализирует скрытый от глаза патологический субстрат.
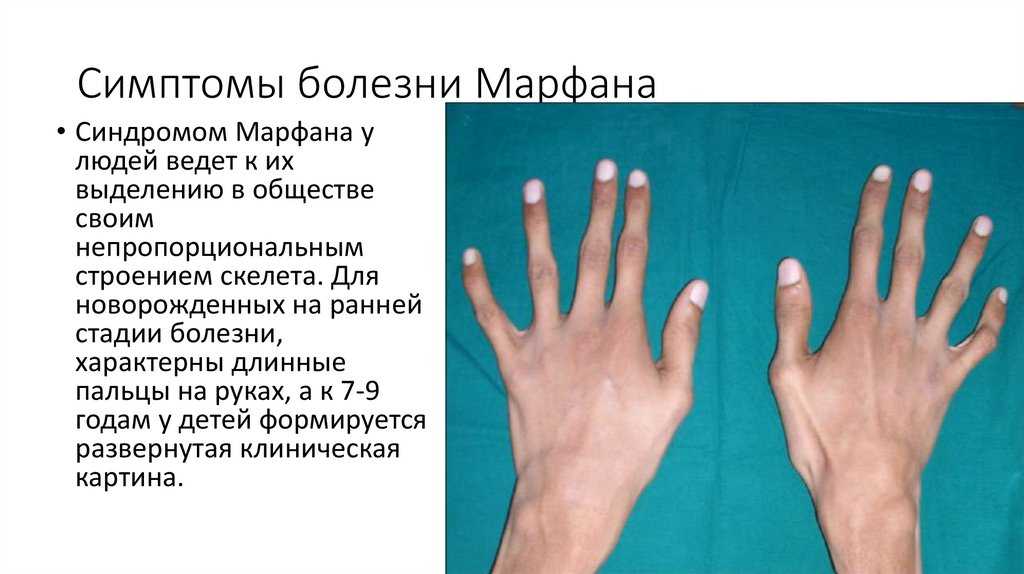
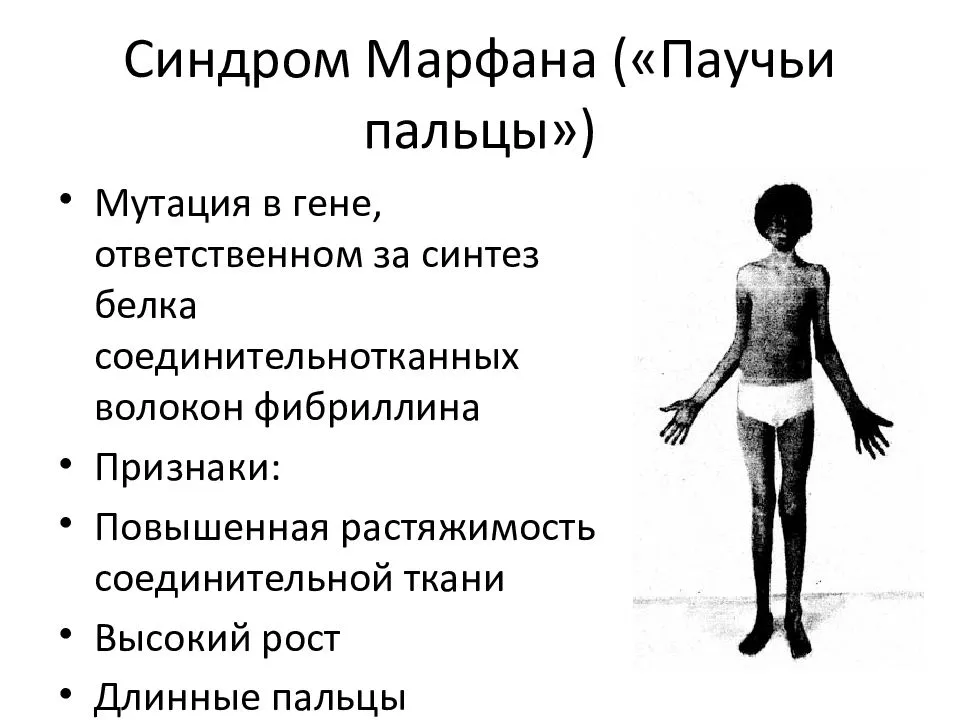
Симптомы синдрома Марфана
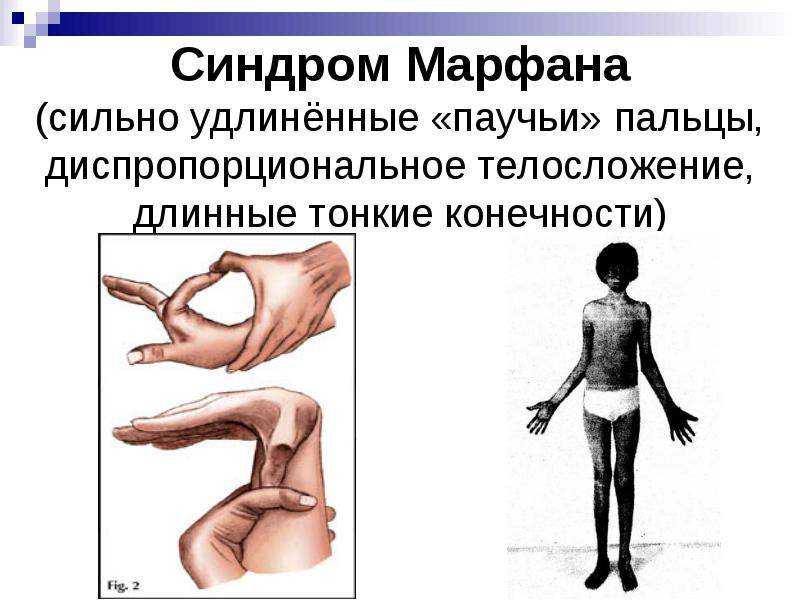
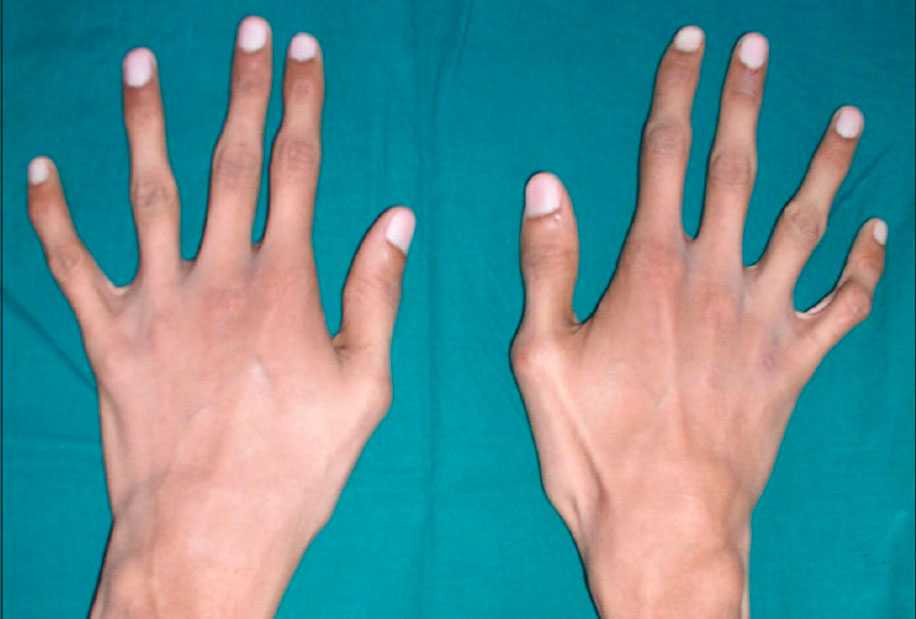
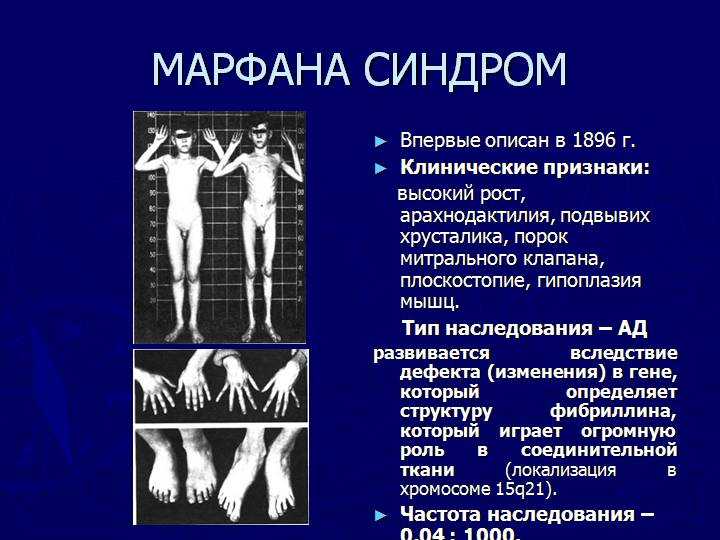
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Восстановительный период
Биопсия шейки матки для диагностики дисплазии при правильном выполнении не чревата проблемами, но некоторым пациенткам с хроническими воспалительными процессами половой сферы требуется профилактическое применение антибиотиков. Не допускается использование в послеоперационном периоде вагинальных тампонов и промывания влагалища, на 2 недели также исключается половая жизнь.
После конизации по поводу дисплазии несколько дней беспокоят не столько боли, сколь неприятные ощущения внизу живота. Будут и выделения, в первые дни могут быть прожилки крови или окрашивание кровью. С течением времени выделения осветляются и уменьшается их количество. Длительность истечения зависит от скорости заживления. Возможно усиление менструации. На полное восстановление слоя слизистой потребуется несколько недель, период ограничений увеличивается, как минимум до месяца, то есть на это время полностью исключается секс, тампоны, спринцевания и высокие физические нагрузки.
Восстановление обязательно должен контролировать врач. Результат лечения оценивает контрольная кольпоскопия и соскоб слизистой – не должно быть признаков атипии и участков дисплазии.
Предраковые процессы и дисплазии умеют лечить обычные гинекологи, но наблюдение специалиста с онкологической подготовкой, тем более в специализированной онкологической клинике поднимает диагностику и терапию на должный уровень качества.
Запись на консультацию круглосуточно
+7 (495) 151-14-53+7 (861) 238-70-54+7 (812) 604-77-928 800 100 14 98
Список литературы
- Каприн А.Д., Новикова Е.Г., Трушина О.И., Грецова О.П. /Скрининг рака шейки матки – нерешенные проблемы // Исследования и практика в медицине; 2015, т. 2, N 1.
- Короленкова Л.И. /Цервикальные траэпителиальные неоплазии и ранние формы рака шейки матки: клинико-морфологическая концепция цервикального канцерогенеза// М., 2017.
- Короленкова Л.И., Ермилова В.Д./ Зона трансформации шейки матки как объект канцерогенного действия вирусов папилломы человека при возникновении CIN и инвазивного рака – отражение в клинике// Архив патологии; 2011, Т. 73, N 6.
- Кудинова Е.Г., Момот А. П. /Наследственные нарушения соединительной ткани и семейный рак: есть ли взаимосвязь?// Архив внутренней медицины; 2015; 4(24).
- Митрофанов А. И., Борзунов Д. Ю. /Результаты лечения пациентов с активными солитарными костными кистами с применением чрескостного остеосинтеза //Гений ортопедии; 2010, № 2.
- Яковлев В.М., Нечаева Г.И., Мартынов А.И., Викторова И.А. /Дисплазия соединительной ткани в практике врачей первичного звена здравоохранения: Руководство для врачей//М.: КСТ Интерфорум; 2016.
- Darragh T.M., Colgan T.J., Cox J.T., et al. /The Lower Anogenital Squamous Terminology Standardization Project for HPV-Associated Lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology // Arch. Pathol. Lab. Med.; 2012 Oct. Vol. 136, N 10.
- Saslow D., Solomon D., Lawson H.W., et al. /American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer // Am. J. Clin. Pathol.; Vol. 137.
Запись на консультацию круглосуточно
+7 (495) 151-14-53+7 (861) 238-70-54+7 (812) 604-77-928 800 100 14 98
Лечение дисплазии
При врожденных дисплазиях радикального лечение не существует, при некоторых опасных для жизни пороках развития проводятся операции, но вся терапия направлена только на уменьшение симптомов с увеличением двигательной активности и паллиативная по своей сути.
При дисплазии суставов разработаны эффективные, но тягостные методики многомесячной фиксации для создания покоя и предоставления времени для не случившегося во внутриутробном периоде развития.
При дисплазии желудка проводится избавление от хеликобактерии, налаживается ритм питания, модифицируется диета и образ жизни в сторону нормального, используются защищающие слизистую от повреждений лекарственные средства.
Лечение дисплазии шейки матки зависит от её выраженности: при легкой степени дисплазии прибегают к консервативным мероприятиям. Вирус папилломы человека устойчив к лекарственному воздействию, но его жизнедеятельность поддерживают нарушения слизистой оболочки при хроническом воспалении, способствует снижение иммунитета при гормональных нарушениях и системных заболеваниях.





Здоровой женщине проще избавится от ВПЧ, но при наличии у неё болезней необходимо помочь организму – излечить острое воспаление, перевести хронический процесс в длительную ремиссию, добиться гормонального баланса, нормализовать сахар и другие элементы крови.
При длительно существующей CIN I, которую наблюдают почти 3 года без тенденции ткани к нормализации структуры, тоже оперируют – удаляют сектор шейки. Сектор похож на геометрическую фигуру – конус, отсюда и название операции – конизация.
Среднюю и тяжелую дисплазию лечат только хирургией – проводится конизация шейки матки радиоволновым или лазерным методами. Операция высокоэффективна, почти не оставляет рубцов и не мешает в дальнейшем забеременеть и выносить ребенка.
Симптомы и клинические проявления
Синдром Марфана может проявляться у разных людей по-разному. У некоторых признаки данного типа патологии выражены очень ярко, у других же наблюдаются исключительно лёгкие симптомы. Обычно прогрессирование симптомов происходит по мере того, как человек растёт. Признаки болезни зависят от того, какие системы и органы в организме поражены, поэтому симптоматика может несколько изменяться.
Синдром Марфана характеризуется:
- сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем;
- многообразием проявлений, варьированием сроков появления первых признаков заболевания;
- хроническим прогредиентным течением.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Вот некоторые основные характеристики людей с синдромом Марфана:
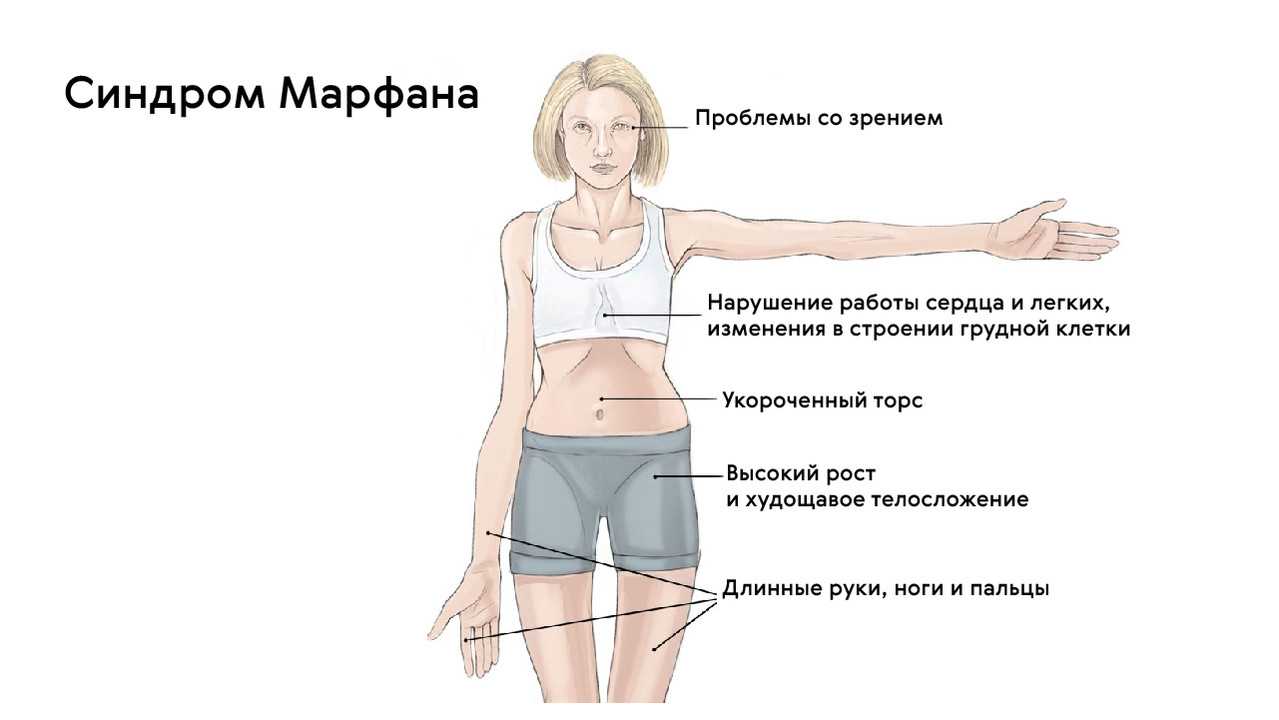
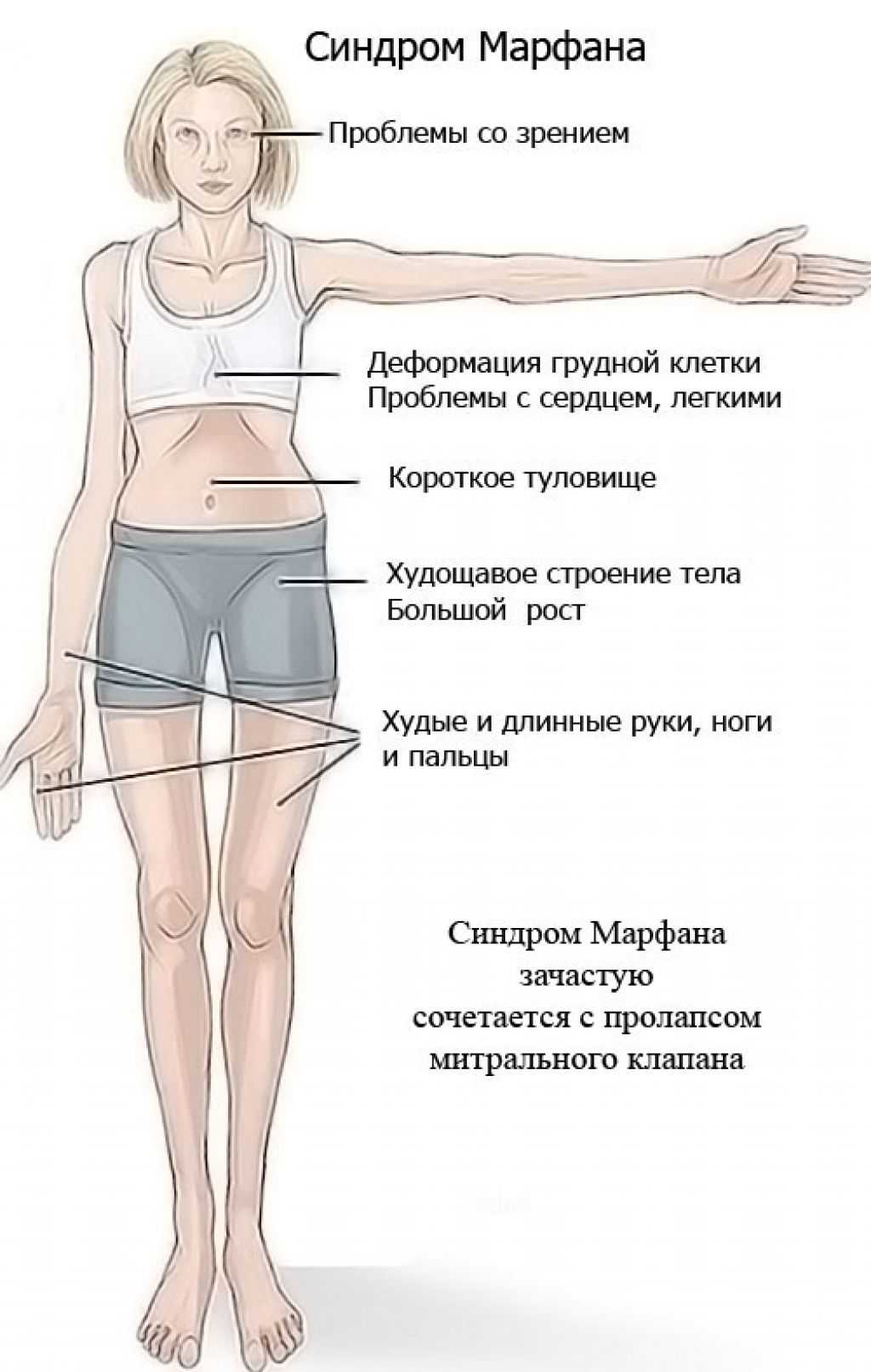
- Скелет — человек с синдромом Марфана обычно очень высокий и худой. В связи с тем, что синдром Марфана сопровождается удлинением костей скелета: туловища, рук, ног, пальцев рук и ног, они могут быть непропорционально длинные. Человек с синдромом Марфана часто имеет длинное, узкое лицо, и его верхняя губа может быть изогнута, причиной чего являются зубы. Другие скелетные аномалии включают изменение грудини (грудной кости), которая или выступает, или зигзагообразной формы, искривление спины (сколиоз) и плоскостопие. Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек — соответственно 52,5 см и 175 см.
- Глаза – у более, чем половины всех людей с синдромом Марфана отмечается смещение одного из двух хрусталиков глаз. Хрусталики глаз могут быть незначительно выше, чем нормальные и смещаться в сторону. Смещение может быть минимальным или резко выраженным и очевидным. Отслоение сетчатки – это серьёзное осложнение. Многие люди с синдромом Марфана близоруки и у них может развиться глаукома (высокое давление внутри глаза) или катаракта.
- Сердце и кровеносные сосуды (сердечно-сосудистая система) – большинство людей с синдромом Марфана имеют аномалии, связанные с сердцем и кровеносными сосудами. Так как имеется дефект соединительной ткани, стенка аорты (большая артерия, которая несет кровь из сердца к телу) может быть ослаблена и растягиваться, этот процесс называют расширением аорты. Расширение аорты — увеличивает риск разрыва аорты, вследствие чего возникает серьёзная проблема с сердцем или иногда внезапная смерть. Иногда могут развиваться дефекты сердечных клапанов. В большинстве случаев, определенные клапаны могут создавать «сердечный шум», который может услышать доктор с помощью стетоскопа. Небольшие нарушения в работе сердца могут не вызывать симптомов, но большие могут стать причиной затруднения дыхания, усталости и сильного сердцебиения (очень быстрого или неправильного сердечного ритма).
- Нервная система – головной и спинной мозг окружены жидкостью; вокруг имеется оболочка, которая называется твердой мозговой оболочкой, ее составляет соединительная ткань. Когда люди с синдромом Марфана стареют, твердая мозговая оболочка часто слабеет и вытягивается. Это называется дуральная эктазия. Эти изменения могут стать причиной легкого дискомфорта или могут привести к боли в брюшной полости или боли, неподвижности или слабости ног.
- Кожа — у большинства людей с синдромом Марфана развивается растягивание кожи, даже без изменения массы тела. Это может появиться в любом возрасте и не представлять опасности. Тем не менее, у людей с синдромом Марфана увеличивается риск развития брюшной или паховой грыжи, при которой развивается выпуклость, которая содержит в себе часть кишки.
- Легкие – хотя аномалии соединительной ткани делают маленькие воздушные мешочки в легких менее эластичным, люди с синдромом Марфана обычно не испытывают существеннных проблем с их легкими. Если, тем не менее, эти маленькие воздушные мешочки удлиняются или распухают, то может увеличиться риск спадения легких. Редко у людей с синдромом Марфана могут возникать нарушения дыхания во время сна, как храп или синдром апноэ (нарушение сна возникает в краткий период, когда дыхание останавливается).
- Самое опасное и скрытое для глаза проявление болезни – это поражение аорты, самой крупной артерии организма, по которой кровь выходит из сердца. У пациентов с синдромом Марфана аорта расширяется и может разорваться. Это может привести к внезапной смерти в молодом возрасте.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на .
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно
Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Симптомы Болезни (синдрома) Марфана:
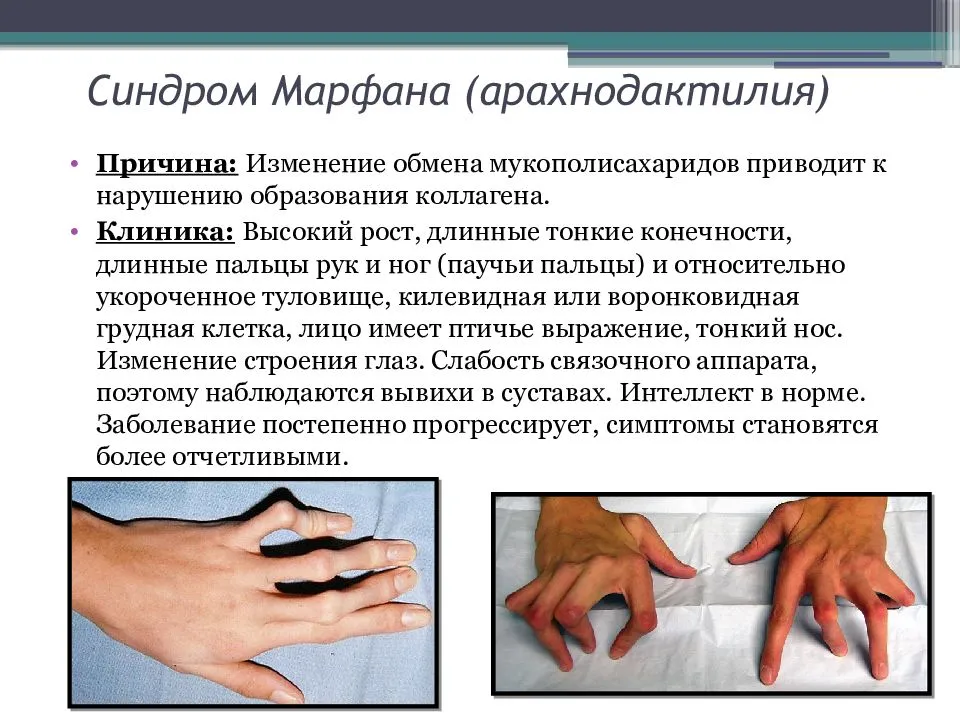
Клинические симптомы заболевания разделяют на несколько групп, которые отражают точную локализацию проявлений соединительнотканной (мезенхимальной) дисплазии:
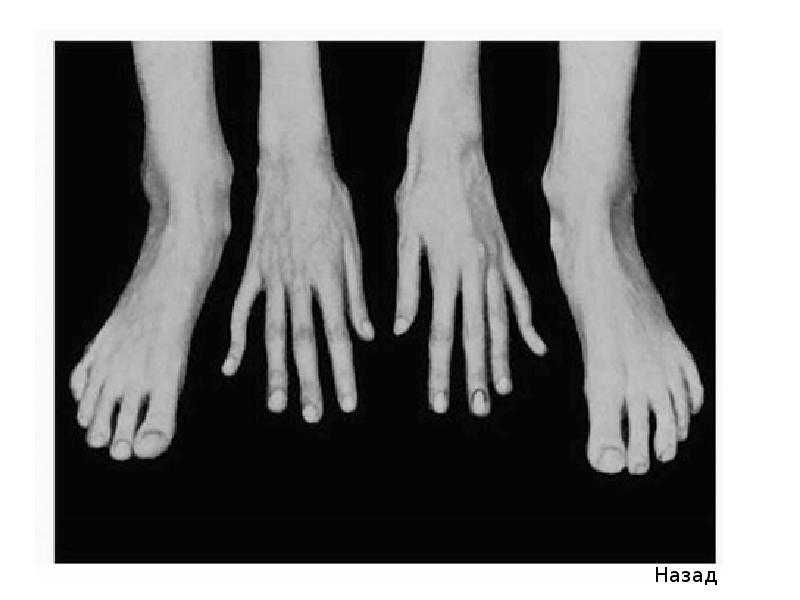
- костно-суставные расстройства (астеническая конституция, узкий лицевой череп с «птичьим» выражением лица, плоскостопие, узкая и деформированная, килевидная или воронкообразная грудная клетка, арахнодактилия кистей и стоп, кифосколиотическая деформация позвоночника, гипермобильность суставов и сухожилий). Костно-суставные нарушения есть у большинства больных;
- изменения мягких тканей (малая масса тела, мышечная гипотония, гипоплазия жировой ткани и мускулатуры, плоскостопие);
- изменения внутренних органов (аневризма восходящей аорты, клапанные пролапсы, особенно пролапс митрального клапана, гипоплазированное, расширение корня аорты и легочной артерии, аневризмы синусов Вальсальвы, «висячее», «капельное» сердце, уменьшение долей легких, слишком длинный и гипопластичный кишечник);
- нарушения в системе зрения (голубые склеры, гиперметропия высоких степеней, аниридия, выраженная миопия, эктопия и подвывихи хрусталика, афакия, колобома);
- расстройства центральной нервной системы (анизокория, нистагм, асимметрия сухожильных рефлексов, пирамидные расстройства);
- Расстройства гипофизарно-адреналовой системы (высокий рост, акромегалоидные расстройства, несахарный диабет, арахнодактилия, удлиненные конечности, увеличенные ступни, вегетативные расстройства);
- нарушения сердечно-сосудистой системы (нарушения внутрижелудочковой проводимости, умеренные признаки гипертрофии миокарда левого желудочка и предсердия, изменения в сердце и магистральных сосудах, аортальная недостаточность, пролапс митрального клапана, нарушения внутрисердечной гемодинамики, расширение корня аорты, двустворчатый клапан аорты, митральная недостаточность, которая связана с развитием миксоматозной дегенерацией створок, увеличением их площади, расширением фиброзного кольца, удлинением хорд, «разболтанностью» створок и увеличением пролабирования). У больных наблюдается мышечная слабость и снижение активности при физических нагрузках.
Как не пропустить синдром Марфана у детей
Поскольку патология имеет наследственную природу, в большинстве случаев удаётся проследить заболевание в семье. Нужно помнить, что недуг может протекать в различных формах и многие люди, с лёгким течение синдрома, не догадываются о своём заболевании на протяжении всей жизни.
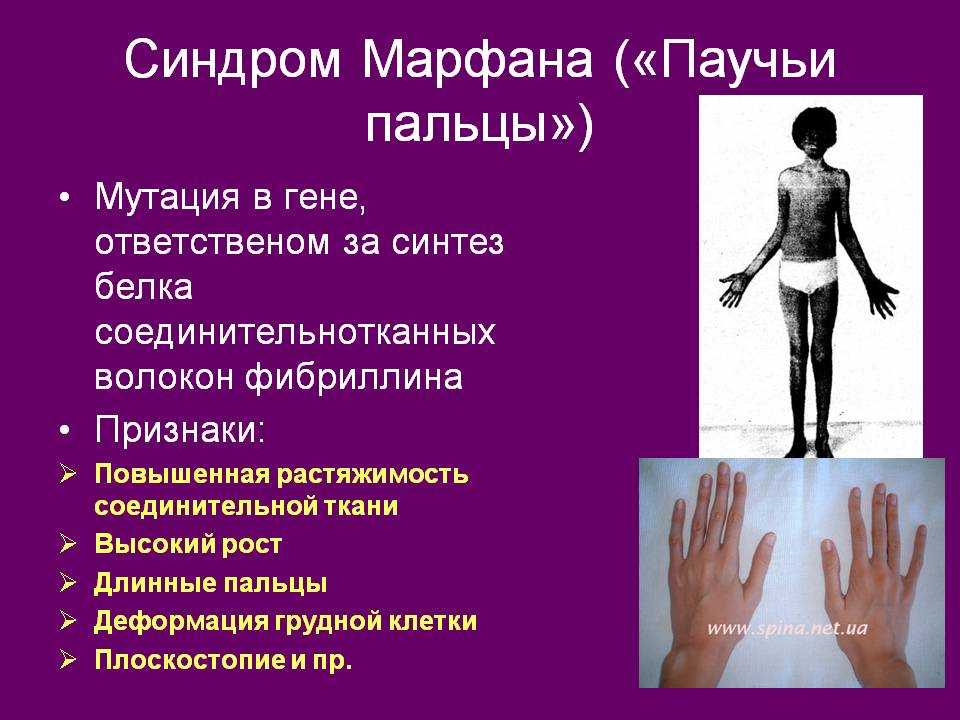

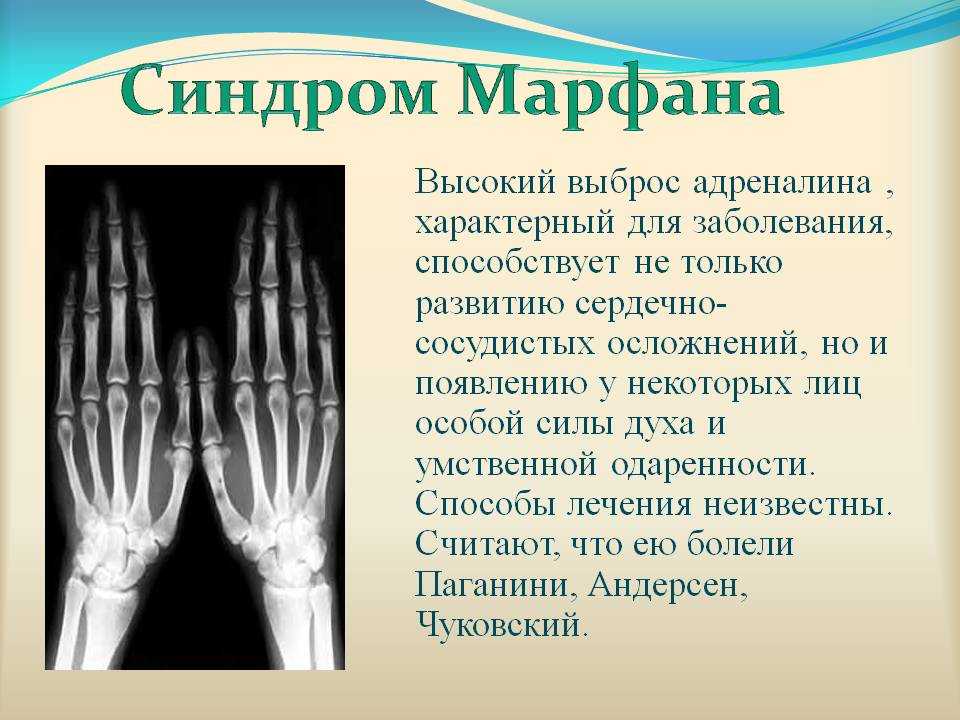
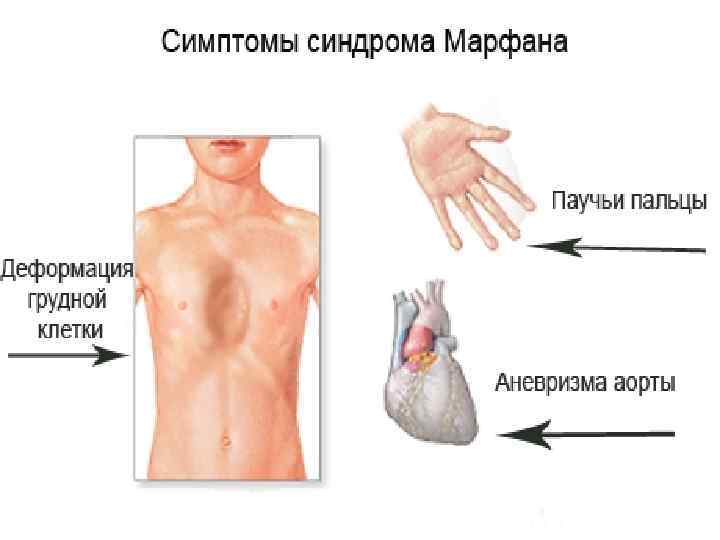


Особенность клинических проявлений
Поскольку болезней, связанных с патологией соединительной ткани, существует множество, иногда бывает сложно правильно поставить диагноз ребёнку. Чтобы справится с этой задачей в 1996 году генетиками и клиницистами были разработаны современные критерии, с помощью которых можно определить синдром Марфана. В основу диагностики были положены «большие» и «малые» признаки заболевания. Их сочетание оценивается специалистом и решается вопрос о наличии генетического синдрома.
Большие критерии
К ним относятся:
- увеличение роста в большей степени за счёт верхней части тела;
- грубая деформация грудной клетки и позвоночника;
- продольное плоскостопие;
- невозможность полностью разогнуть конечность в коленных и локтевых суставах (контрактуры);
- эктопия хрусталика;
- расширение и расслоение восходящей части аорты и другие признаки.
Некоторые из этих симптомов можно встретить у абсолютно здоровых детей. В диагностике генетического синдрома большую роль играет именно их сочетание.
Триада Марфана включает в себя:
- патологию костно-суставной системы,
- органические изменения в сердце или крупных сосудах,
- болезни глаз.
Малые критерии
Эти признаки в меньшей степени указывают на наследственный дефект, но их наличие и сочетание с большими критериями подтверждает диагноз синдром Марфана.
К ним относятся:
- высокая подвижность суставов;
- аномалии зубов, нёба;
- гипоплазия радужной оболочки глаза, цилиарной мышцы, увеличение длины глазного яблока;
- пролапс митрального клапана;
- патологии бронхо-лёгочной системы,
- спонтанный пневмоторакс и другие нарушения.
Диагностические тесты
Для уточнения внешних изменений у ребёнка используются различные способы: измерение роста, длины кисти, соотношение размеров верхней части туловища к нижней и другие. Особо показательны в диагностике синдрома следующие исследования:
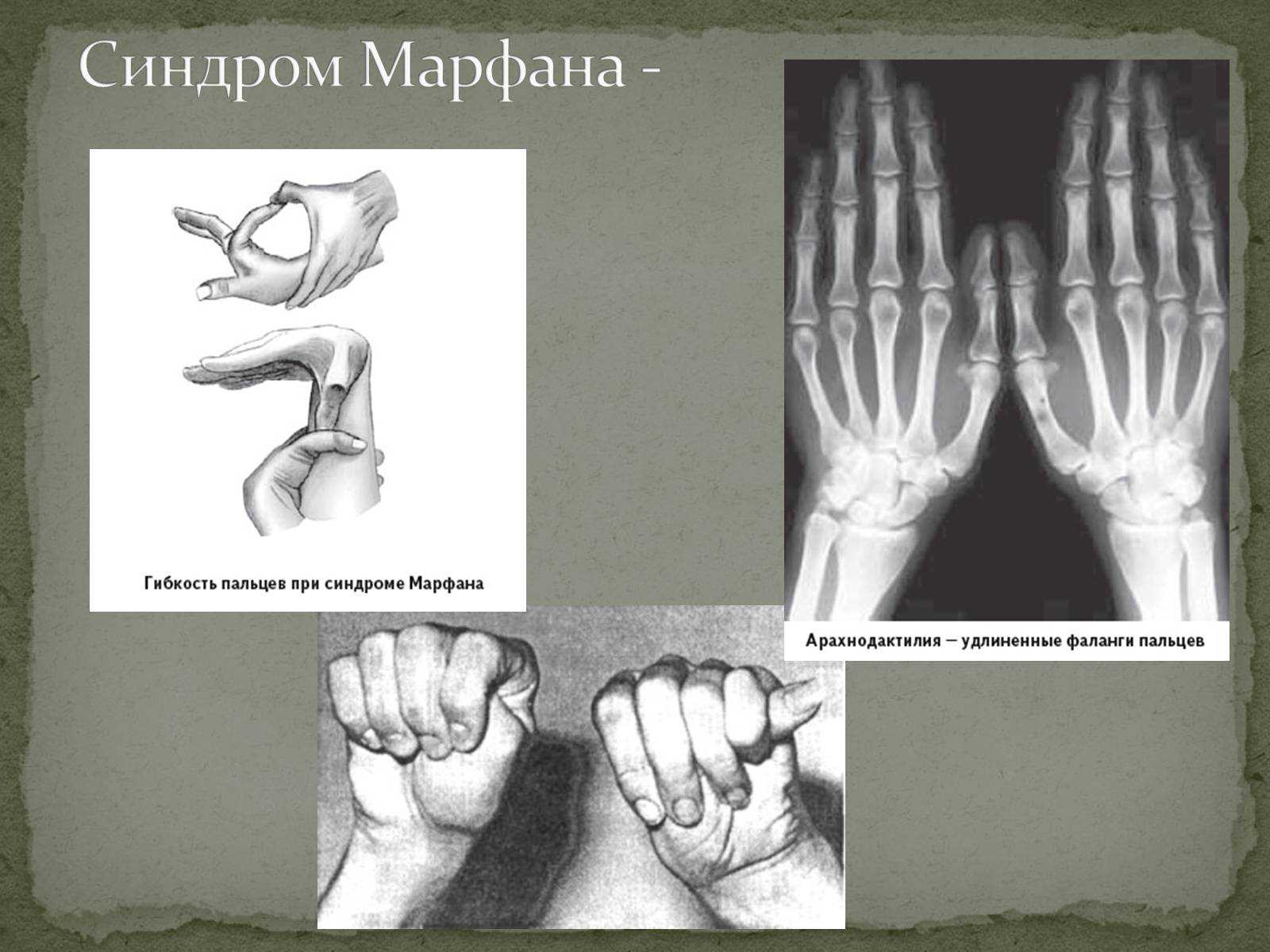
- тест запястья. Врач просит ребёнка обхватить запястье одной руки большим пальцем и мизинцем другой руки, образуя «браслет». В пользу наследственного недуга говорит лёгкое смыкание кисти на запястье другой руки, нахождение фаланг мизинца и большого пальца друг на друга;
- тест большого пальца. Исследователь просит малыша попытаться дотянутся большим пальцем до предплечья этой же руки. Тест считается положительным, если ногтевая фаланга пальца ребёнок с лёгкостью достаёт до лучевой кости предплечья.
Лабораторные исследования
Обычные клинические и биохимические анализы крови и мочи не показательны при синдроме Марфана, изменений в них может не быть. Помочь в установлении диагноза поможет обнаружение продуктов метаболизма соединительной ткани в моче. Резкое увеличение оксипролина и гликозаминогликанов в суточной моче может говорить о развитии осложнений у ребёнка (прогрессировании сердечной недостаточности, отслойке плаценты, развитии пневмоторакса, пневмонии).
С помощью современных методов исследования, молекулярно-генетической диагностики можно обнаружить характерную для данного генетического синдрома мутацию в гене FBN1.
Такое заболевание, как синдром Билса отличается от синдрома Марфана дефектом синтеза другого белка – фибриллина 2, но клинические симптомы этих недугов схожи. Различить их можно только с помощью молеклярно-генетической диагностики и выявления особого симптома «мятого уха», что указывает на наличие синдрома Билса.
Общие данные
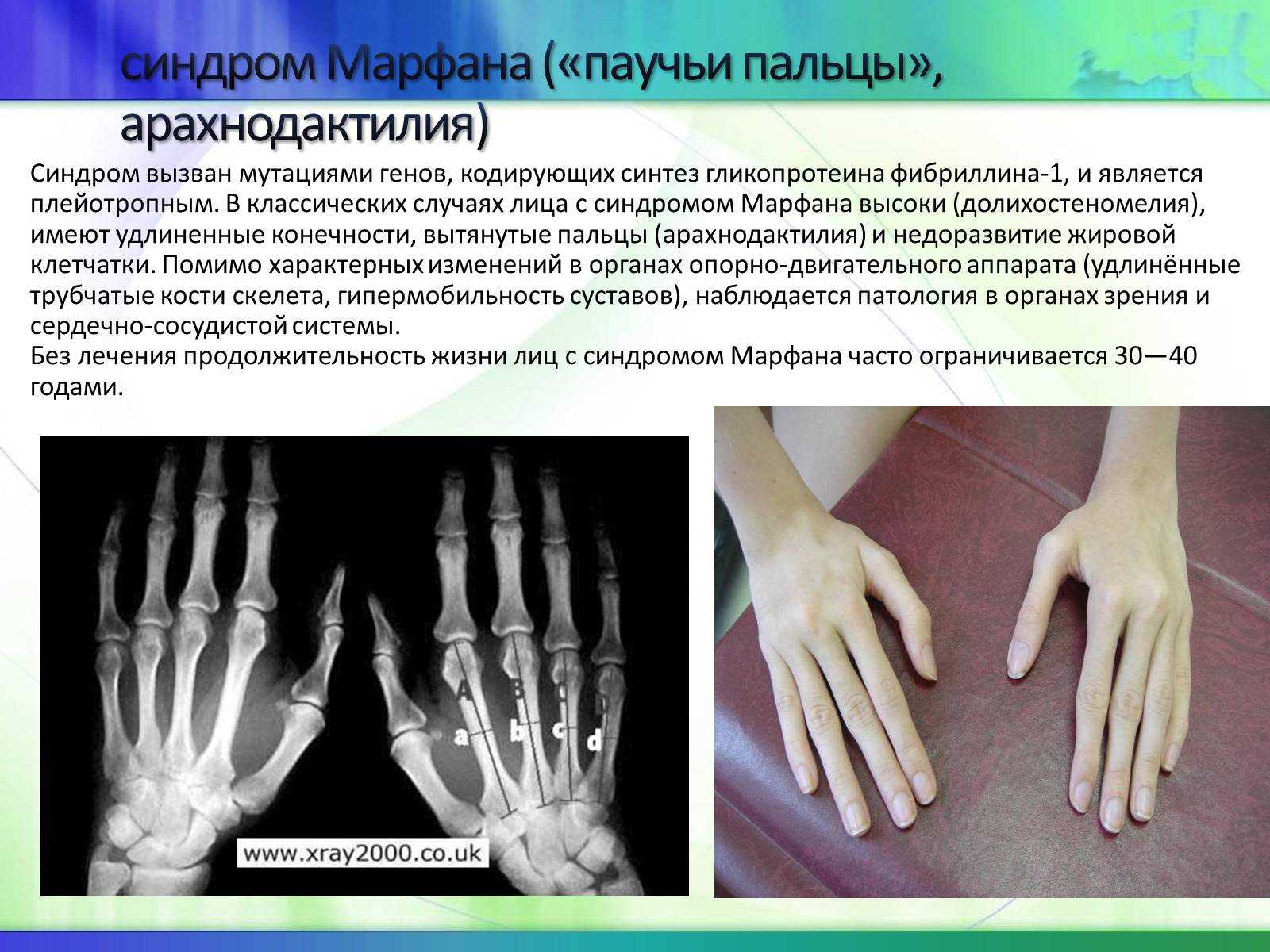
Синдром был открыт в 1875 году офтальмологом из Америки Э. Вильямсом. Он обнаружил полное смещение хрусталика глаза от своего нормального положения у брата и сестры, которые имели высокий рост и чрезмерную подвижность суставов с рождения. Спустя несколько лет детский врач из Франции А. Марфан вел наблюдения за девочкой 5 лет с прогрессирующими аномалиями скелета, чрезмерно длинными конечностями и «паучьими пальцами». Он дал четкое описание патологии, благодаря чему синдром получил его имя.
Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.
По данным американского Национального Фонда Синдрома Марфана, в США проживает около 200 000 человек с этим заболеванием, и лишь половина из них диагностированы. Представители фонда активно информируют общественность о признаках этого заболевания, чтобы люди могли обратиться за медицинской помощью, ведь это заболевание встречается гораздо чаще, чем многие думают.
Синдром Марфана развивается вследствие дефекта (изменения) в гене, который определяет структуру фибрина, который играет огромную роль в соединительной ткани. Человек с синдромом Марфана рожден с нарушением, даже если это не было установлено в течение жизни. Хотя каждый человек с синдромом Марфана имеет дефект определенного гена, изменения специфичны для каждой семьи и не каждый может испытывать подобные симптомы в одинаковой степени. Это называют подразумевает, что дефектный ген проявляется у различных людей в различной степени.
Дефектный ген может быть унаследован: ребенок имеет 50% вероятность унаследовать заболевание от родителя с синдромом Марфана. Иногда новый генный деффект появляется во время соединения сперматозоида и яйцеклетки, здоровые родители имеют один шанс из 10,000, родить ребенка с синдромом Марфана. Возможно, в 25% случаев происходит самопроизвольное изменение во время оплодотворения.
Патологии сетчатки при синдроме Марфана
Из-за слабости соединительной ткани подвержена растяжению также сетчатка глаза. В результате этого повышается риск появления периферических хориоретинальных дистрофий — локальных истончений сетчатки, которые могут спровоцировать отторжение слоя светочувствительных клеток от пигментного эпителия. Такое нарушение очень опасно: начинает падать острота зрения, ухудшается восприятие света и цвета.
Признаками отслойки сетчатки могут быть следующие проявления:
- вспышки, искры в глазах — такое явление называется фотопсией;
- искажение формы, размера и оттенка объектов — метаморфопсия;
- «мушки» и черные точки перед глазами как следствие повреждения ретинального сосуда;
- выпадение из поля зрения отдельных элементов в видимой картинке — это признак того, что отслоение началось в центральной зоне сетчатки;
- появление темной пелены, охватывающей все большую область, снижение периферийной видимости.

Отслоение сетчатки сегодня поддается успешному лечению различными методами. Наиболее эффективным из них является лазерная коагуляция — прижигание поврежденных участков с целью надежного их соединения с сосудистой оболочкой.

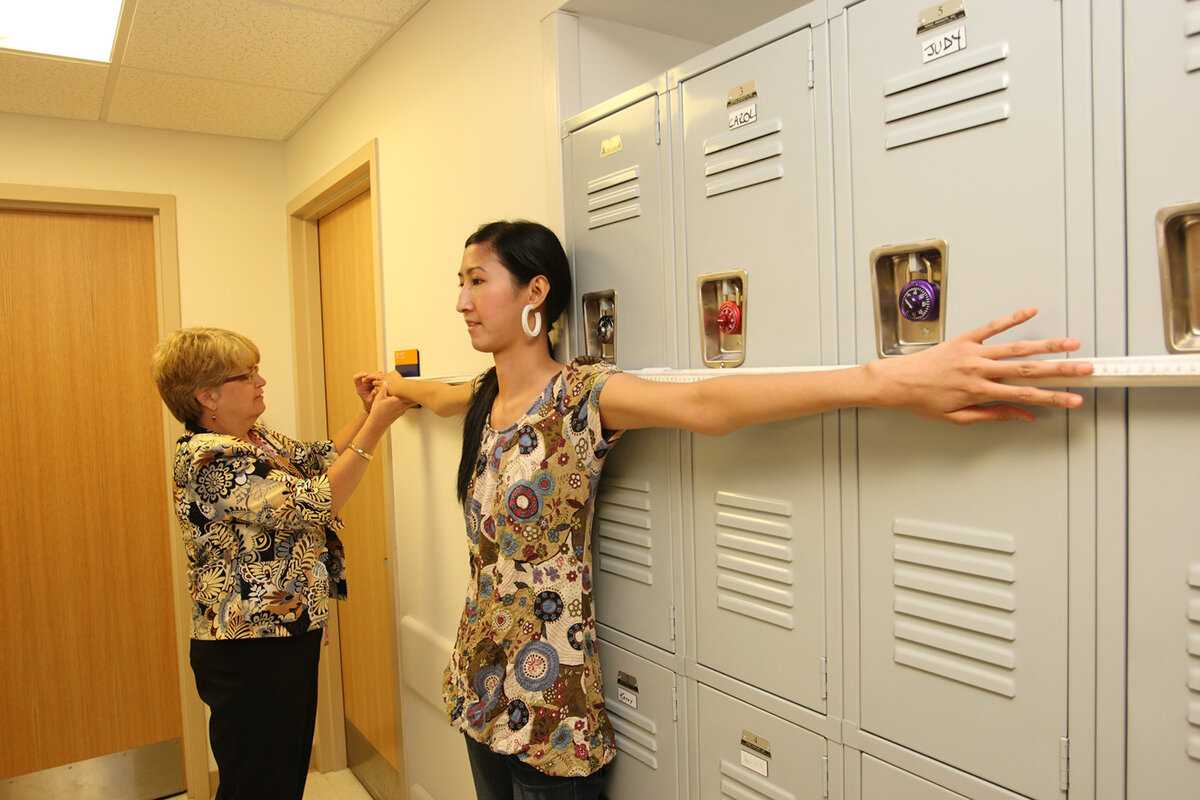




Наследование синдрома Марфана в наши дни
Врожденный синдром Марфана передается по аутосомно–доминантному типу. Это значит, что его появление в семье подчиняется таким закономерностям:
- чаще болен один из родителей;
- частота появления у девочек и мальчиков одинаковая;
- вероятность возникновения у ребенка равна 50%;
- от здоровых родителей (при наличии больных бабушек, дедушек) обычно не передается;
- если провести анализ родословного древа, то выявляется вертикальный способ передачи синдрома в каждом поколении;
- не все члены семьи имеют одинаковые признаки болезни.
Следует учитывать, что в 20% случаев мутации гена не связаны с наследственностью, они могут возникать первично. В таких случаях играет роль возраст родителей. Доказано, что чаще дети с этой аномалией развития появляются, если отец старше 35 лет. В следующем поколении больных при таком варианте наследования может и не быть (пропуск поколения), но половина их потомков окажется с этой патологией. Поэтому всем будущим родителям, у которых в семьях были выявлены лица с синдромом Марфана, нужно перед планированием беременности обязательно пройти консультацию в медико-генетическом центре.



